
|
第一章 细胞、组织的适应和损伤
|
适应
细胞、组织的损伤
萎缩
原因
肥大
发生机制
增生
形态学变化:变性、坏死、凋亡
化生
―――――――――――――――――――――――――――――――――――――
细胞是人体的基本结构单位。细胞的生命活动是在内、外环境的动态平衡过程中进行的。
细胞和由其构成的组织、器官能耐受内、外环境中各种有害因子的刺激作用而得以存活的过程,称为适应(adaptation)。适应在形态上表现为萎缩、肥大、增生和化生。
细胞和组织遭受不能耐受的有害因子刺激时,则可能引起损伤(injury)。较轻的细胞损伤是可逆的,即消除刺激因子后,受损伤细胞可恢复常态,通常称之为变性或是亚致死性细胞损伤(sublethal
cell injury)。这后一名称或许较“变性”一词更确切些,但“变性”已习惯引用。细胞的严重损伤是不可逆的,最终引起细胞死亡。
细胞和组织的适应性和损伤性变化是疾病发生的基础性病理变化。
第一节
适应
一、萎缩
萎缩(atrophy)是指已发育正常的实质细胞、组织或器官的体积缩小,可以伴发细胞数量的减少。组织、器官的实质细胞萎缩时,常继发其间质(主要是脂肪组织)增生,有时使组织、器官的体积甚至比正常还大,称为假性肥大(见于萎缩的胸腺、肌肉等)。器官先天地部分性和完全性未发育所致的体积小,分别称为发育不全(hypoplasia)和不发育(agenesis),并非萎缩。萎缩细胞的蛋白质合成减少而分解增多,以适应其营养水平低下的生存环境。
萎缩分为生理性和病理性两类。人体的许多组织、器官,例如胸腺、生殖系统等,随年龄增长自然地发生生理性萎缩。病理性萎缩按其发生原因可分为:①
营养不良性萎缩,例如脑动脉粥样硬化时因慢性缺血所致的脑萎缩;蛋白质等摄入不足或消耗过多引起的全身性营养不良性萎缩,如饥饿、慢性消耗性疾病,和恶性肿瘤的恶病质等;② 压迫性萎缩,例如尿路梗阻时,因肾盂积水引起的肾萎缩(图1-1);③
废用性萎缩,因长期工作负荷减少所致的萎缩,例如久卧不动时的肌肉萎缩、骨质疏松;④
去神经性萎缩,例如因神经、脑或脊髓损伤所致的肌肉萎缩;⑤
内分泌性萎缩,例如因腺垂体肿瘤或缺血性坏死等引发的肾上腺萎缩,严重者还可致甲状腺、性腺和全身性萎缩(Simmonds综合症)。心肌、脑等的老年性萎缩兼有生理性和病理性萎缩。
轻度病理性萎缩时,去除原因后,萎缩的细胞有可能恢复常态;持续性萎缩的细胞终于死亡。
萎缩细胞胞浆内的细胞器大量退化、自噬小体增多,因而可有大量未能被溶酶体酶降解、富含磷脂的残体(即脂褐素)积聚,常见于心肌、肝细胞和神经节细胞(参见脂褐素沉着)。
萎缩的器官均匀性缩小,重量减轻,功能低下。萎缩的心脏因心肌胞浆中脂褐素沉着而呈褐色(褐色萎缩)。大脑萎缩时,脑回变窄,脑沟变深,皮质变薄,体积缩小,重量变轻(参见图13-18)。镜下见神经细胞体积缩小、数量减少。
图1-1
二、肥大
细胞、组织和器官体积的增大,称为肥大(hypertrophy)。肥大分为生理性肥大(physiologic
hypertrophy)和病理性肥大(pathologic
hypertrophy)。细胞肥大通常具有功能代偿意义,多属于代偿性肥大(compensatory
hypertrophy)。由激素引发的肥大称为内分泌性肥大(endocrine
hypertrophy)。肥大的组织、器官常伴发细胞数量的增多(增生),即肥大常与增生并存。骨骼肌和心肌是不具分裂能力的永久性细胞,只能以代偿性肥大适应其工作负荷的增加,例如运动员有关肌肉的生理性肥大,高血压时左心室排血阻力增加所致的左心室肌壁病理性肥大(参见图6-7)。妊娠期子宫和哺乳期乳腺发生生理性肥大常兼有增生,属于内分泌性(激素性)肥大。
细胞的肥大导致由其组成的组织和器官体积增大、重量增加和功能增强。因代偿而肥大的器官超过其代偿限度时便会失代偿(decompensation),例如肥大心肌的失代偿引发心力衰竭。
三、增生
实质细胞的增多称为增生(hyperplasia),增生可导致组织、器官的增大。细胞增生时也常伴发细胞肥大。受机体调控的细胞增生,随其有关引发因素的去除而停止。这显然不同于肿瘤细胞的失控性增生。但是,过度增生的细胞有可能演变为肿瘤性增生。
细胞增生常与激素和生长因子的作用有关。生理和病理情况下都可发生激素性增生,例如女性青春期乳腺和妊娠期的子宫均属生理性增生;雌激素水平升高所致的子宫内膜和乳腺增生则属病理性增生。功能代偿也可引发增生,例如低钙血症引发的甲状旁腺代偿性增生。
细胞增生通常为弥漫性,以致增生的组织、器官弥漫、均匀地增大。在有关激素的过度作用下,前列腺、甲状腺、肾上腺和乳腺等常呈结节性增生。这可能是由于这类器官中有的靶细胞对于激素的作用更为敏感,因而在正常或大致正常的组织中形成单个或多发性结节。
四、化生
一种分化成熟的细胞因受刺激因素的作用转化为另一种分化成熟细胞的过程称为化生(metaplasia)。化生主要发生于上皮细胞,也见于间叶细胞,可能与干细胞(如上皮组织的贮备细胞,间叶组织的原始间叶细胞)调控分化的基因重新编程(reprogramming)有关,属于细胞的转型性分化(transdifferentiation)。这种分化上的转向通常只发生于同源性细胞之间,即上皮细胞之间和间叶细胞之间。化生有多种类型,最常为柱状上皮(例如子宫颈管和支气管粘膜的腺上皮)、移行上皮等化生为鳞状上皮,称为鳞状上皮化生(简称鳞化)(图1-2)。萎缩性胃炎时胃粘膜腺上皮的肠上皮化生(简称肠化)(参见图8-3)。在间叶组织中,纤维组织可化生为软骨组织或骨组织(例如骨化性肌炎时的骨组织形成)。化生的生物学意义利害兼有,以呼吸道粘膜纤毛柱状上皮的鳞化为例,化生的鳞状上皮一定程度地强化了局部抗御环境因子刺激的能力,因此属于适应性变化,但是,却减弱了粘膜的自净机制。化生的上皮可以恶变,如由被覆腺上皮的粘膜(例如肺内的支气管粘膜)可发生鳞状细胞癌,胃粘膜可发生肠型腺癌。
图1-2
第二节
细胞、组织的损伤
一、原因
引发细胞损伤的原因很多,可以归纳为:缺氧、化学物质和药物、物理因素(机械性、高低温、高低气压、电流、射线等)、生物因子(细菌、病毒、真菌、寄生虫等)、营养失衡(过剩或缺乏)、内分泌因素、免疫反应、遗传变异、衰老、社会-心理-精神因素和医源性因素等若干大类。
缺氧是许多致病因素引起细胞损伤的一个非常重要的基本环节。老年人体的细胞处于生理性自然退化状态,对于长期积累下来的或是即时接受的各种致病因素的损伤作用会更为敏感,从而导致衰老(aging)或老化(senility)。
不良的社会-心理-精神刺激是现代社会中日益受到重视的致病因素,由这种思想、情感障碍引发细胞损伤所形成的器质性疾病称为心身疾病(psychosomatic
disease)。例如心理-精神障碍是原发性高血压、消化性溃疡、冠心病和植物神经功能紊乱等的一个重要发病因素,甚至也可成为潜在恶性肿瘤临床发作的重要因素。
在对患者原有疾病进行诊断、治疗时,由于诊治过程本身继发的伤害属于医源性疾病(iatrogenic
disease)。医生在临床工作中要注意防范。
二、发生机制
由多种因素引发的细胞损伤,其发生机制是很复杂的。细胞膜的破坏、活性氧类物质(氧自由基等)增多、胞浆内高游离钙、缺氧、化学毒作用和遗传物质变异等,对于细胞损伤的发生具有重要意义。这几方面的机制常是相互结合或是互为因果地导致细胞的损伤。
(一)细胞膜的破坏
细胞膜是由脂质(70%为磷脂)和蛋白质组成的,是细胞与外部环境之间的界膜或屏障,是保持细胞生命活动的基本结构。细胞以细胞膜与外界互通信息、交换物质,在免疫应答、细胞分裂和分化等方面也具有重要作用。细胞内、外多种有害因素可以破坏细胞膜的结构和功能,从而导致细胞的损伤。可以破坏细胞膜的因素包括机械力的直接作用、脂酶性溶解、缺氧和活性氧、补体结合、感染、药物性损伤等。
(二)活性氧类物质的损伤作用
活性氧类物质(activated
oxygen species,AOS)或称反应性氧类物质(reactive oxygen species,ROS),包括处于自由基状态的氧和不属于自由基的过氧化氢(H2O2),前者包括超氧自由基(O2)和羟自由基(.OH)。自由基(free
radicals)是原子最外层偶数电子失去一个电子后形成的具有强氧化活性的基团。AOS以其对于脂质、蛋白质和DNA的氧化作用损伤细胞。细胞内同时存在生成AOS体系和拮抗其生成的抗氧化剂体系。因此,正常时少量生成的AOS会及时被抗氧化剂(例如超氧歧化酶)清除。细胞在多种致病因素作用下,AOS生成增多,导致细胞损伤。AOS的强氧化作用,是细胞损伤发生机制的基本环节。
(三)细胞浆内高游离钙的损伤作用
细胞浆内的磷脂酶和内切核酸酶等能降解磷脂、蛋白质、ATP和DNA。这两种酶的作用需要游离钙活化。胞浆内游离钙与ATP依赖性钙转运蛋白结合,成为蛋白结合钙并贮存于线粒体、内质网等钙库内。正常时,胞浆因此处于低游离钙状态,使上述酶类活性稳定,细胞的结构和功能得以保持。细胞膜内依赖于ATP的钙泵和钙离子通道也参与胞浆内游离钙浓度的调节,使胞浆内游离钙减少。缺氧、中毒等致使ATP减少时,细胞的胞浆内会因此继发游离钙增多,上述的酶类因而活化,使细胞损伤。胞浆内高游离钙所引发的酶活化是多种致病因素导致细胞损伤发生机制的终末环节。
(四)缺氧的损伤作用
缺氧(hypoxia)是指细胞不能获得足够氧或是氧利用障碍。缺氧是一个很常见的重要病理过程。缺氧的原因,可以是:①
空气中氧分压低或气道(外呼吸)障碍,属于单纯性缺氧;②
血红蛋白的质、量异常,属于血液性缺氧;③
局部性缺血或心、肺功能衰竭,属于循环性缺氧;④
直接中毒所致线粒体内呼吸(生物氧化,主要是氧化磷酸化)障碍,属于细胞中毒性缺氧。其实,上述前三种原因引发的缺氧最终都导致线粒体氧化磷酸化受抑制,使ATP生成减少,造成细胞膜钠-钾泵、钙泵功能低下和胞浆内蛋白质合成、脂肪代谢(脂肪运出胞浆)障碍等,造成无氧糖酵解过程的活化。酵解会使细胞酸中毒、溶酶体膜破裂,并损伤核染色质(DNA链)。缺氧还可使氧自由基等活性氧类物质增多,膜磷脂丢失,脂质崩解,细胞骨架破坏等。轻度、较短时间缺氧所致的细胞损伤通常是可逆的(神经细胞除外),一般引发细胞的水肿、脂肪变;严重缺氧或/和较长时间的轻度缺氧所致的细胞损伤是不可逆的,引发细胞坏死。图1-3以局部缺血为例,说明缺氧所致细胞损伤的发生机制。
(五)化学性损伤
化学性损伤(chemical
injury)包括化学物质和药物的毒性作用,日益成为致细胞损伤的重要因素。作为医治疾病的药物具有可能引发细胞损伤的副作用,是最常见的医源性致病因子。
化学性损伤可表现为:①
全身性:例如氰化物中毒;②
局部性:例如吞食强酸、强碱所致口腔、食管和胃粘膜的腐蚀;③
器官特异性:例如有机磷、四氯化碳引发的肝损伤。
影响化学性损伤的重要因素包括:①
剂量:与损伤程度正相关,小剂量的毒性作用与其持续作用的时间正相关;②
吸收、蓄积、代谢或排出的部位:例如链霉素、庆大霉素、丁胺卡那霉素等因蓄积于内耳的内、外淋巴和肾小管上皮细胞而分别具有耳毒性和肾毒性;③
代谢速度的个体差异:与有关酶活性的遗传性差异有关。
化学性物质和药物损伤细胞的途径包括:①
直接的细胞毒性作用:例如氰化物因迅速封闭线粒体的细胞色素氧化酶系统而致猝死。②
代谢产物对于靶细胞的细胞毒性作用:肝、肾、心肌、骨髓等是这种毒性代谢产物的主要靶器官。肝细胞是许多化学物质和药物,特别是有机磷农药或杀虫剂、四氯化碳、酒精、细胞毒性抗肿瘤药等的代谢部位,尤易遭受毒性代谢产物的损伤,通常表现为中毒性或药物诱发性肝炎;③
诱发免疫性损伤:例如青霉素可经由Ⅰ型变态反应引发过敏反应,氯霉素可经由Ⅱ型变态反应引发粒细胞减少症、再生障碍性贫血等;④诱发DNA损伤(见下文“遗传变异”)
(六)遗传变异
化学物质和药物、病毒、射线等可损伤细胞核内的DNA,诱发基因突变和染色体畸变,使细胞发生遗传变异(genetic
variation),可导致:① 结构蛋白合成低下:细胞可因缺乏生命必需蛋白质而死亡;②
核分裂受阻:正常时核分裂活跃的骨髓造血干细胞、肠粘膜上皮细胞和睾丸精母细胞等的生理性增生因而低下,分别引发粒细胞缺乏或再生障碍性贫血、小肠吸收功能障碍和男性不育症等;③
合成异常生长调节蛋白:例如转化性蛋白质等可诱发单克隆转化性细胞形成,进而形成肿瘤;④
酶合成障碍:引发先天性代谢病或后天性酶缺陷,细胞可因缺乏生命必需的代谢机制发生死亡。
![]()
![]()
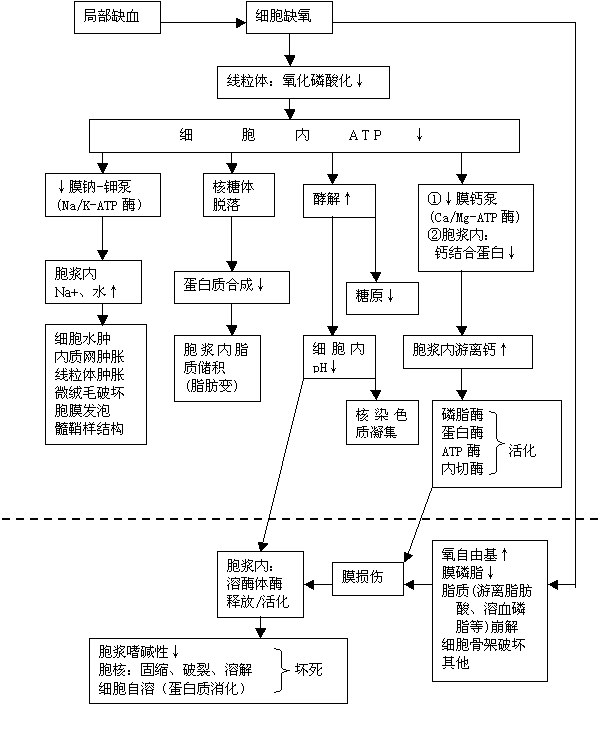
图1-3
细胞缺血-缺氧性损伤的发生机制
三、细胞、组织损伤的形态学变化
以缺氧为例,遭受了损伤的细胞,首先表现为代谢性变化(图1-3);经过一段不等的时间(例如缺血后数分钟至数小时),受损伤细胞呈现组织化学和超微结构的变化,然后(例如缺血后数小时至数日),才呈现光镜下和用肉眼可见的形态学变化,即病理变化或病变(lesions)。这种损伤性病变包括:①
在去除病因后有可能恢复常态的可逆性病变,亚致死性细胞损伤或变性;②严重的不可逆性病变即细胞死亡。
(一)变性
变性(degeneration)是指细胞或细胞间质受损伤后因代谢发生障碍所致的某些可逆性形态学变化。表现为细胞浆内或细胞间质内有各种异常物质或是异常增多的正常物质的蓄积,每伴有功能下降。
1.细胞水肿(cellular
swelling)
或称为水变性(hydropic
degeneration),是细胞轻度损伤后常发生的早期病变,好发于肝、心、肾等实质细胞的胞浆。细胞水肿的主要原因是缺氧、感染和中毒。其发生机制是:缺氧时线粒体受损伤,使ATP生成减少,细胞膜Na+-K+泵功能因而发生障碍,导致胞浆内Na+、水增多。
病理变化:电镜下,胞核正常,胞浆内的线粒体、内质网等肿胀呈囊泡状(图1-4)。光镜下,弥漫性细胞胀大,胞浆淡染、清亮,核可稍大,重度水肿的细胞称为气球样变(见于病毒性肝炎)(图1-5)。肉眼观,发生了细胞水肿的肝、肾等体积增大、颜色变淡。去除病因后,水肿的细胞可恢复正常。
图1-4
图1-5
2.脂肪变
细胞浆内甘油三酯(中性脂肪)的蓄积称为脂肪变(fatty
change)或脂肪变性(fatty
degeneration)。起因于营养障碍、感染、中毒和缺氧等。多发生于肝细胞、心肌纤维和肾小管上皮。
病理变化:电镜下,细胞胞浆内的脂肪表现为脂肪小体,进而融合成脂滴。光镜下,于HE染片中,脂滴表现为大小不等的近圆形空泡(因脂肪被制片时的有机溶剂溶解之故)(图1-6);于冷冻切片中,蓄积于胞浆内的脂肪可用脂溶性的苏丹Ⅲ染料染成红色。去除病因后,蓄积于胞浆内的脂肪可消失。
肝细胞是脂代谢的部位,最常发生脂肪变。显著弥漫性肝脂肪变称为脂肪肝。肉眼观:肝增大、边缘钝、色淡黄、较软,切面油腻感。重度脂肪变的肝细胞,其胞核被胞浆内蓄积的脂肪压向一侧,形似脂肪细胞,并可彼此融合成大小不等的脂囊(图1-6)。肝细胞脂肪变通常不引起肝功能障碍。重度脂肪变的肝细胞可坏死,并可继发肝硬化。
图1-6
肝细胞脂肪变的机制
血液中的脂肪酸进入肝细胞胞浆后,经由下列过程进行代谢:①
被氧化为酮体和CO2;②
参与磷脂和胆固醇的合成;③
在a-磷酸甘油的参与下合成甘油三酯(中性脂肪),甘油三酯经与载脂蛋白结合以脂蛋白的形式进入血流。肝细胞脂肪酸代谢过程的某个或多个环节,由于下列因素的作用而异常时,便可引发脂肪变:①
肝细胞胞浆内脂肪酸增多:高脂饮食或身体皮下、大网膜等处脂肪组织大量分解(例如营养不良时)可致血液脂肪酸增多;机体缺氧所致肝细胞糖酵解过程生成的乳酸可转化为多量脂肪酸;肝细胞内脂肪酸也可因其氧化过程低下而相对增多。②
饮酒:可致a-磷酸甘油增多而促进甘油三酯合成。③
缺氧、营养不良(蛋白质缺乏、饥饿、糖尿病等)和肝毒物质(CCl4等)使载脂蛋白减少,进而脂蛋白减少,甘油三酯蓄积于肝细胞胞浆内。
心肌脂肪变常累及左心室的内膜下和乳头肌,肉眼上表现为大致横行的黄色条纹,与未脂肪变的暗红色心肌相间,形似虎皮斑纹,称为虎斑心。
心外膜处显著增多的脂肪组织,可沿心肌层的间质向着心腔方向伸入,心肌因受伸入脂肪组织的挤压而萎缩并显薄弱,称为心肌脂肪浸润(fatty
infiltration),并非脂肪变性。重度心肌脂肪浸润时,浸润于心肌内的脂肪组织可接近(甚至达于)心内膜下方,可导致心肌破裂、出血,引发猝死。
3.玻璃样变
玻璃样变(hyaline
change)又称玻璃样变性或透明变性(hyaline
degeneration),泛指细胞内、纤维结缔组织间质或细动脉壁等,在HE染片中呈现均质、粉染至红染、毛玻璃样半透明的蛋白质蓄积。其发生机制各异。
(1)细胞内玻璃样变:蓄积于细胞内的异常蛋白质形成均质、红染的近圆形小体,通常位于细胞浆内。例如,肾小管上皮细胞的玻璃样小滴变性(蛋白尿时由原尿中重吸收的蛋白质)(图1-7)、浆细胞胞浆中的Russell小体(蓄积的免疫球蛋白)和酒精性肝病时肝细胞胞浆中的Mallory小体(参见图8-31)等。
图1-7
(2)纤维结缔组织玻璃样变:是胶原纤维老化的表现,见于纤维结缔组织的生理性增生,例如发生于萎缩的子宫、乳腺、睾丸等和病理性增生,例如瘢痕、动脉粥样硬化斑块、肾小球纤维化、硅肺、心瓣膜病、浆膜粘连、血栓或坏死组织的机化等。镜下:增生的胶原纤维变粗、融合,形成均质、粉色或淡红染的索、片状结构,其中很少纤维细胞和血管(图1-8、1-11,参见图10-19)。肉眼观:大范围透明变性的纤维结缔组织(例如大块瘢痕)呈灰白色、均质半透明,较硬韧。胶原纤维透明变性可能是由于胶原蛋白交联增多,使胶原纤维大量融合、多量糖蛋白蓄积其间;也可能是胶原蛋白变性、融合的结果。
图1-8
(3)细动脉壁玻璃样变:又称细动脉硬化(arteriolosclerosis),常见于缓进性高血压和糖尿病患者,弥漫地累及肾、脑、脾和视网膜等处的细小动脉壁。玻璃样变的细小动脉壁因有蛋白质蓄积而显增厚、均质性红染,管腔狭窄,可导致血管变硬,血液循环外周阻力增加和局部缺血;管壁弹性减弱、脆性增加,因而继发扩张,导致破裂出血(图1-9)。
图1-9
4.淀粉样变
淀粉样变(amyloidosis)是在细胞外的间质内,特别是小血管基底膜处,有蛋白质-粘多糖复合物蓄积,并显示淀粉样呈色反应,即遇碘液后呈棕褐色,再遇稀硫酸时由棕褐色变为深蓝色。这种淀粉样物质(amyloid)在HE染片中呈均质性粉色至淡红色,类似玻璃样变(图1-10),但被刚果红染成红色、甲基紫染成紫红色。电镜下,淀粉样物质呈细丝状(宽约0.75~10nm)。局部性淀粉样变发生于皮肤、眼结膜、舌、喉、气管和肺、膀胱、胰岛(糖尿病时)等处,也可蓄积于恶性淋巴瘤和神经内分泌肿瘤(例如甲状腺髓样癌)的间质内。全身性淀粉样变分为原发性和继发性。继发性者的淀粉样物质来源未明,常继发于严重的慢性炎症,例如慢性空洞性肺结核病,慢性化脓性骨髓炎等和某些恶性肿瘤;原发性者的淀粉样物质来源于免疫球蛋白的轻链。全身性淀粉样变时可累及许多部位,引发相关的临床表现。肝、脾、肾、心常受累及,体积增大,色泽较淡,质地较脆,可因其实质细胞被压萎缩而发生功能障碍。
图1-10
5.粘液样变性
粘液样变性(mucoid
degeneration)是指间质内有粘多糖(透明质酸等)和蛋白质的蓄积。常见于间叶组织肿瘤、风湿病、动脉粥样硬化和营养不良时的骨髓和脂肪组织等。镜下:间质疏松,有多突起的星芒状纤维细胞散在于灰蓝色粘液样基质中。甲状腺功能低下时,可能是由于甲状腺素减少所致的透明质酸酶活性减弱,使含有透明质酸的粘液样物质以及水分蓄积于皮肤及皮下的间质中,形成粘液性水肿(myxedema)。
6.
病理性色素沉着
有色物质(色素)在细胞内、外的异常蓄积称为病理性色素沉着(pathologic
pigmentation)。沉着的色素主要是由体内生成的(内源性色素),包括含铁血黄素、脂褐素、胆红素、黑色素等。随空气吸入肺内的炭尘和纹身时注入皮内的着色物质等属于外源性色素沉着。
(1)含铁血黄素(hemosiderin):组织内出血时,从血管中逸出的红细胞被巨噬细胞摄入并由其溶酶体降解。使来自红细胞血红蛋白的Fe3+与蛋白质结合成电镜下可见的铁蛋白微粒,若干铁蛋白微粒聚集成为光镜下可见的棕黄色、较粗大的折光颗粒,称为含铁血黄素。巨噬细胞破裂后,此色素也可见于细胞外。含铁血黄素因含Fe3+被普鲁氏蓝染成蓝色。生理情况下,红细胞在肝、脾内破坏,可有少量含铁血黄素形成。含铁血黄素的病理性沉着多为局部性,提示陈旧性出血。当溶血性贫血时,有大量红细胞被破坏,可出现全身性含铁血黄素沉积,主要见于肝、脾、淋巴结和骨髓等器官。
(2)脂褐素(lipofuscin):是蓄积于胞浆内的黄褐色微细颗粒,电镜下显示为自噬溶酶体内未被消化的细胞器碎片残体,其中50%为脂质。附睾管上皮细胞、睾丸间质细胞和神经节细胞的胞浆内正常时便含有脂褐素。老年人及一些慢性消耗性疾病患者的心肌细胞、肝细胞、肾上腺皮质网状带细胞等萎缩时,其胞浆内有多量脂褐素沉着。故此色素又有消耗性色素之称。
(3)黑色素(melanin):是由黑色素细胞生成的黑褐色微细颗粒。在腺垂体分泌的ACTH和MSH(黑色素细胞刺激激素)促进下,黑色素细胞胞浆中的酪氨酸在酪氨酸酶的作用下,经由多巴(dihydroxyphenylalanine,DOPA)反应生成黑色素。局部性黑色素增多见于色素痣、恶性黑色素瘤等。肾上腺皮质功能低下的Addison病患者可呈现全身性皮肤、粘膜的黑色素沉着。这是因为肾上腺皮质激素分泌减少,对垂体的反馈抑制减弱,致使ACTH和MSH分泌增多之故。
7.病理性钙化
在骨和牙齿外的软组织内有固体性钙盐(主要是磷酸钙和碳酸钙)的沉积称为病理性钙化(pathologic
calcification)。在HE染色时,钙盐呈蓝色颗粒状~片块状。继发于局部变性、坏死组织或其他异物(例如血栓、死亡的寄生虫卵)内的钙化,称为营养不良性钙化(dystrophic
calcification)(图1-11)。营养不良性钙化时,体内钙磷代谢正常。由于钙磷代谢障碍(高血钙)所致正常肾小管、肺泡、胃粘膜等处的多发性钙化,称为转移性钙化(metastatic
calcification),可影响细胞、组织的功能。甲状旁腺功能亢进、骨肿瘤破坏骨组织、维生素D过多摄入等可引发高血钙,导致转移性钙化。
图1-11
(二)细胞死亡
细胞因受严重损伤而累及胞核时,呈现代谢停止、结构破坏和功能丧失等不可逆性变化,此即细胞死亡(cell
death)。细胞死亡包括坏死和凋亡两大类型。
1.坏死
坏死(necrosis)是活体内范围不等的局部细胞死亡,死亡细胞的质膜(细胞膜、细胞器膜等)崩解、结构自溶(坏死细胞被自身的溶酶体酶消化)并引发急性炎症反应。炎症时渗出的嗜中性粒细胞释放溶酶体酶,可促进坏死的发生和溶解。
坏死可迅即发生,也可由可逆性损伤(变性)发展而来。
(1)基本病变:细胞死亡几小时(例如心肌梗死后4~12小时)后,光镜下才可见坏死细胞开始呈现自溶性变化。胞核一般依序呈现
① 核固缩(pyknosis):表现为核缩小、凝聚,呈深蓝染,提示DNA停止转录;② 核碎裂(karyorrhexis):表现为染色质崩解成致密蓝染的碎屑,散在于胞浆中,核膜溶解;③ 核溶解(karyolysis):染色质中的DNA和核蛋白被DNA酶和蛋白酶分解,核淡染,只见、甚至不见核的轮廓(图1-12、1-14)。胞浆红染,胞膜破裂,坏死细胞进而解体、消失;间质内胶原纤维肿胀、崩解、液化,基质解聚。最后坏死的细胞和崩解的间质融合成一片模糊的无结构的颗粒状红染物质。
坏死初发时,首先呈现琥珀酸脱氢酶、乳酸脱氢酶等的活性降低(可用酶组织化学染色显示),进而呈现超微结构变化。电镜下,坏死细胞胞核的染色质最初高度致密并聚集于胞膜附近(边集),细胞器退变,线粒体肿大、破裂和钙盐沉积,溶酶体也肿大、破裂,致使坏死细胞自溶(图1-13)。
由于坏死细胞膜通透性增加,胞浆中的一些酶可释放至血液中(浓度升高),临床上可借以作为诊断某些部位细胞坏死性疾病的参考指标,例如心肌梗死时的血液肌酸激酶、乳酸脱氢酶、谷草转氨酶升高;肝细胞坏死时的血液谷草转氨酶、谷丙转氨酶升高;胰腺坏死时的血液淀粉酶升高等。
图1-12
图1-13
(2)类型:坏死分为凝固性坏死、液化性坏死和纤维素样坏死三个基本类型,前两种坏死又有一些特殊类型。
1)凝固性坏死(coagulative
necrosis):坏死细胞的蛋白质凝固,还常保持其轮廓残影(图1-14)。这可能是由于坏死局部的酸中毒使坏死细胞的结构蛋白和酶蛋白变性,封闭了蛋白质的溶解过程。凝固性坏死好发于心肌、肝、脾、肾等。
干酪样坏死(caseous
necrosis)是彻底的凝固性坏死,是结核病的特征性病变。镜下:不见坏死部位原有组织结构的残影,甚至不见核碎屑(图1-15);肉眼观:坏死呈白色或微黄,细腻,形似奶酪(参见图14-4),因而得名。
坏疽(gangrene)是身体内直接或间接地与外界大气相通部位的较大范围坏死,并因有腐败菌生长而继发腐败。坏疽分为干性、湿性和气性三种。干性和湿性坏疽多继发于动脉阻塞引起的缺血性坏死(梗死)。肉眼观呈黑色,其与坏死局部来自红细胞血红蛋白的Fe2+与蛋白质分解出的H2S形成硫化铁有关,边界清楚。干性坏疽(dry gangrene)常继发于肢体,首先是肢端等水分容易蒸发的体表组织坏死,由于坏死组织干燥,腐败菌感染一般较轻(图1-16)。湿性坏疽(moist
gangrene)常继发于肠管、胆囊、子宫、肺等与外界沟通,但水分不易蒸发的脏器坏死,也可继发于动脉受阻同时有静脉淤血的体表组织坏死,由于坏死组织含水分较多,腐败菌感染严重。气性坏疽(gas
gangrene)常继发于深在的开放性创伤,特别是战伤,合并厌氧的产气荚膜杆菌等感染时,细菌分解坏死组织产生大量气体,使坏死组织内含气泡呈蜂窝状。湿性坏疽,尤其是气性坏疽可伴发全身性中毒。
图1-14
图1-15
图1-16
2)液化性坏死(liquefactive
necrosis):是坏死组织因酶性分解而变为液态。最常发生于含可凝固的蛋白少和脂质多的脑和脊髓,又称为软化(malacia)(参见图13-6)。化脓、脂肪坏死和由细胞水肿发展而来的溶解性坏死(lytic
necrosis)都属于液化性坏死。脂肪坏死(fat
necrosis)包括创伤性和酶解性两大类。创伤性者好发于皮下脂肪组织(尤其是女性乳房),致脂肪细胞破裂,脂肪外溢,引起巨噬细胞和异物巨细胞吞噬脂质反应,局部形成肿块;酶解性者见于急性胰腺炎,与胰脂酶外溢消化胰周脂肪组织有关。镜下,坏死脂肪细胞仅留下模糊混浊的轮廓(图1-17)。脂肪坏死时因有大量脂肪酸形成常继发营养不良性钙化(钙皂形成)。肉眼见为白色的斑点或斑块。
图1-17
3)纤维素样坏死(fibrinoid
necrosis):曾称为纤维素样变性,发生于结缔组织和血管壁,是变态反应性结缔组织病(风湿病、类风湿性关节炎,系统性红斑狼疮、结节性多动脉炎等)和急进性高血压的特征性病变。镜下,坏死组织呈细丝、颗粒状的红染的纤维素(纤维蛋白)样,聚集成片块(图1-18)。纤维素样坏死物质可能是肿胀、崩解的胶原纤维(由于抗原-抗体复合物引发),或是沉积于结缔组织中的免疫球蛋白,也可能是由血液中渗出的纤维蛋白原转变成的纤维素。可能由于疾病的不同,纤维素样物质的成分也有异。
图1-18
(3)结局
1)细胞坏死后发生自溶,并在坏死局部引发急性炎症反应。
2)坏死组织溶解,经由淋巴管、血管吸收,或被巨噬细胞吞噬清除。小范围坏死可被完全吸收、清除。较大范围坏死液化后可形成囊腔(cyst)。
3)坏死组织分离、排出,形成缺损。皮肤、粘膜处的浅表性坏死性缺损称为糜烂(erosion),较深的坏死性缺损称为溃疡(ulcer),由于坏死形成的开口于表面的深在性盲管称为窦道(sinus),两端开口的通道样坏死性缺损称为瘘管(fistula)。在有天然管道与外界相通器官(例如肺、肾等),较大块坏死组织经溶解后由管道(支气管-口腔、输尿管-尿道)排出后残留的空腔,称为空洞(cavity)。
4)机化(organization)。坏死物不能完全溶解吸收或分离派出,则由新生的肉芽组织吸收、取代坏死物的过程称为机化。最终形成瘢痕组织。
5)包裹(encapisulation)。坏死灶如较大,或坏死物质难于溶解吸收,或不完全机化,最初则由肉芽组织包裹,以后则为增生的纤维组织包裹。
6)坏死组织可继发营养不良性钙化。
机体内的异物如血栓,如不能发生溶解吸收,也可发生机化和钙化。
(4)后果
坏死对机体的影响,与下列因素有关:①
坏死细胞的生理重要性,例如心肌、脑组织的坏死后果严重;②
坏死细胞的数量,例如肝细胞的广泛性坏死后果严重;③
坏死细胞所在器官的再生能力,例如肝细胞易于再生,坏死后容易恢复;④
发生坏死器官的贮备代偿能力,例如肾、肺为成对的器官,贮备代偿能力强,即便有较大的坏死也不会明显的影响功能。
2.凋亡
凋亡(apoptosis)是活体内单个细胞或小团细胞的死亡,死亡细胞的质膜(细胞膜和细胞器膜)不破裂,不引发死亡细胞的自溶,也不引起急性炎症反应。凋亡的发生与基因调节有关,也有人称之为程序性细胞死亡(programmed
cell death,PCD)。凋亡不仅与胚胎发生、发展、个体形成、器官的细胞平衡稳定等有密切的关系,并在人类肿瘤、自身免疫性疾病、病毒性疾病等的发生上具有重要意义。
电镜下,凋亡的细胞皱缩,质膜完整,胞浆致密,细胞器密集、不同程度退变;核染色质致密,形成形状不一、大小不等的团块边集于核膜处,进而胞核裂解、胞浆多发性芽突;胞浆芽突迅速脱落,形成许多凋亡小体。凋亡小体外被以胞膜,其胞浆中含有细胞器,核碎片可有可无(图1-19)。凋亡小体迅即在局部被巨噬细胞和相邻的其他细胞(例如上皮细胞)吞噬、降解。细胞凋亡和细胞坏死的超微形态比较参见图1-20。光镜下,凋亡小体多呈圆形或卵圆形,大小不等,胞浆浓缩,强嗜酸性,可有可无固缩深染的核碎片,故有称之为嗜酸性小体。病毒性肝炎中所见的嗜酸性小体实为肝细胞的凋亡(图8-23)。
图1-19
图1-20
(张乃鑫)
参考文献
1.
Stevens A, Lows J. Pathology.London: Mosby, 1995.9-33.
2.
Chadrasoma P, Taylor CR. Concise Pathology.2nd ed. London: Prentill-Hall
International, 1995.1-19.
3.
Underwood JCE. General and Systematic Pathology. Edinburgh:
Churchill Livingstone, 1992.71-77.
4.
董郡.
病理学(中国医学百科全书).
上海:上海科学技术出版社,
1986.31-60.
5.
Damijanow I. Histopathology, A Color Atalas and Textbook.
Baltimore: Williams & Wilkins, 1996.1-22.
6.
Wheater PR, Burkitt HG, Steven A, et al. Basic Histopathology, A
Colour Atlas and Text. Edinburgh: Churchill Livingstone, 1991.2-7.
7.
Sandritter
W, Thomas C. Color Atlas and Textbook.Chicago: Year Book Medical
Publisher, 1979.1-17.
8. Goran ADT,Macfarlane PS,Callander R. Pathology Illustrated (ELBS). Singapore:Churchill Livingstone,1991.2-7.